Giuseppe Curciarello, Coordinatore Team Terapie Domiciliari AIL-Firenze, Ambulatorio specialistico Ematologia e Medicina Trasfusionale, Firenze
I medici di Medicina Generale (MMG), e direi tutti i medici, ben conoscono le alterazioni nel protidogramma e le componenti monoclonali e così i medici delle Strutture Trasfusionali (ST) che da ormai diversi anni hanno precise indicazioni per la sospensione dei donatori con queste alterazioni. La segnalazione del Laboratorio di Patologia Clinica della presenza di una “componente omogenea” in zona gamma o talvolta, anche se meno frequentemente, in altre sedi del tracciato elettroforetico proteico costituisce infatti il segno ematochimico della presenza di un clone plasmacellulare abnorme che appunto produce questa immunoglobulina (Ig). Vedremo in questa disamina come studiare questa alterazione e quali patologie può sottendere e non solo il Mieloma Multiplo.
Mi preme subito ricordare che solo l’immunofissazione sierica permette di confermare la presenza di una componente monoclonale al tracciato elettroforetico delle proteine e che le segnalazioni indicate dai laboratoristi nel commento al protidogramma, esame importante nella valutazione dello stato di salute, sono alla fine il trigger per eseguire, in primis, una immunofissazione sierica che confermi la presenza della Ig monoclonale e la caratterizzi.
Quindi sicuramente la segnalazione da parte del Laboratorio di “alterazione nel tracciato” ovvero di “componente omogenea in zona gamma” o in altra zona del tracciato o la segnalazione, talvolta franca da parte del laboratorio di “sospetta componente monoclonale”, devono indurre il medico ospedaliero, il MMG o il medico della ST a richiedere l’Immunofissazione sierica.
Solo la conferma della presenza di una componente monoclonale, con indicazione della Ig coinvolta (catena pesante e leggera), dovrà indurre il medico curante all’esecuzione di accertamenti e studio della componente monoclonale riscontrata.
Le componenti monoclonali proteiche sono prodotte, come indica il termine “monoclonale”, da un clone di plasmacellule, le cellule adibite alla produzione delle immunoglobuline.
Si parla di “discrasie plasmacellulari”, alterazioni nella proliferazione, appunto clonale, di plasmacellule che secernono una immunoglobulina e raramente anche due immunoglobuline monoclonali.
Si tratta dunque di una alterazione caratterizzata dalla secrezione di proteine evidenziabili nella elettroforesi (protidogramma), immunologicamente omogenee (monoclonali), che esprimono molecole di immunoglobulina completa o talvolta incompleta. Le proteine monoclonali sono comunemente indicate come proteine M o “paraproteine” e in genere mettono in allarme il medico curante (MMG o medico della ST) in quanto sono note come tipiche di una malattia importante quale è il mieloma multiplo (MM).
Molte sono le sindromi associate alle discrasie plasmacellulari con proteine monoclonali ed esse sono indicate con il termine di Gammapatie monoclonali di significato indeterminato (MGUS), MM smouldering, MM sintomatico, macroglobulinemia di Waldenström, nonché disturbi principalmente correlati alle proprietà della proteina monoclonale secreta (crioglobulinemia, amiloidosi AL, malattia da deposito di catene leggere e altre condizioni).
MA VEDIAMO COSA SONO LE IMMUNOGLOBULINE SIERICHE
Le immunoglobuline complete sono costituite da due catene polipeptidiche pesanti (H) della stessa classe e sottoclasse e due catene polipeptidiche leggere (L) dello stesso tipo. Le catene polipeptidiche pesanti sono designate da lettere greche: γ in immunoglobulina G (IgG), α in immunoglobulina A (IgA), µ in immunoglobulina M (IgM), δ in immunoglobulina D (IgD) e ε in immunoglobulina E (IgE). I tipi di catena leggera possono essere o kappa (κ) o lambda (λ). Sia le catene pesanti che le catene leggere hanno regioni “costanti” e “variabili” rispetto alla sequenza aminoacidica. La specificità di classe di ogni immunoglobulina è definita da una serie di determinanti antigenici sulle regioni costanti delle catene pesanti (γ, α, µ, δ e ε) e le due classi principali di catene leggere (κ e λ). La sequenza aminoacidica nelle regioni variabili della molecola di immunoglobulina corrisponde al sito attivo di combinazione con l’anticorpo. Nella maggior parte dei disturbi delle plasmacellule, le molecole di immunoglobuline intatte sono secrete come proteine monoclonali (M). Inoltre, ci può anche essere una secrezione anormale di catene leggere libere in eccesso (definite FLC da Free Light Chains) che vengono rilasciate senza essere legate a catene pesanti di immunoglobuline. D’altra parte dobbiamo immaginare la produzione delle Ig come una sorta di “fabbrica” in cui si costruiscono nella cellula catene pesanti e leggere che poi vengono assemblate, ma c’è sempre un eccesso nella produzione di catene leggere e questo condiziona la non infrequente presenza di catene leggere nelle urine, quando quest’ultime superano la capacità di riassorbimento renale. Queste catene leggere se sono però di origine non clonale, saranno sempre kappa e lambda insieme, cioè miste. Ecco perché, talvolta nell’esame delle urine può comparire ed essere segnalata la presenza di catene kappa e lambda. Queste catene esprimono però solamente una proteinuria ma non una proteinuria cosiddetta di Bence Jones che invece è caratterizzata sempre e solo dalla presenza di una sola catena leggera (appunto espressione della monoclonalità) o kappa o lambda. In alcuni pazienti, la plasmacellula clonale perde capacità di costruire le catene pesanti e vengono secrete solo catene leggere libere monoclonali che superano il tubulo renale (proteinuria di Bence Jones). Meno frequentemente, vengono secrete solo catene pesanti, con la conseguente malattia detta appunto della catena pesante (HCD).
Per completezza dobbiamo anche aggiungere come in rari pazienti con Mieloma multiplo le plasmacellule non secernono immunoglobuline identificabili (si parla di “mieloma non secernente”).
COME IDENTIFICARE LE COMPONENTI MONOCLONALI
L’elettroforesi proteica del siero e dell’urina rileva la proteina M.
L’elettroforesi (Figura 1) consente anche la quantificazione delle proteine M. Le catene leggere monoclonali (proteine di Bence Jones) sono facilmente rilevabili all’elettroforesi delle urine. Inizialmente sarà sufficiente anche solo la ricerca della cosiddetta Bence Jones qualitativa su un semplice campione di urine, ma una raccolta delle urine delle 24 ore si renderà necessaria già come primo step se la componente M è quantitativamente importante ovvero se presente una positività al test Bence Jones qualitativo. Sarà infatti importante nello studio della malattia e anche successivamente durante la terapia monitorare l’effettiva perdita di catena leggera nelle 24h e valutare la riduzione quantitativa.
Quando si osserva per la prima volta un picco o una banda sull’elettroforesi proteica, deve essere dunque eseguita l’immunofissazione del siero e delle urine per identificare il tipo di catena pesante e leggera della proteina M. L’immunofissazione può essere eseguita insieme all’elettroforesi quando si sospetta la presenza di una componente monoclonale oppure, sulla base di segni e sintomi, un mieloma multiplo, e in questi casi l’immunofissazione è particolarmente importante così come lo è nel mieloma oligosecernente, nell’amiloidosi primaria (AL) e nel plasmacitoma solitario, nonché dopo le terapie eseguite.
Le proteine monoclonali devono essere distinte da un eccesso di immunoglobuline policlonali (uno o più tipi di catena pesante e catene leggere κ e λ, di solito nella regione γ), che producono nel tracciato una “cupola” a banda larga e non un picco a base stretta. Questa ipergammaglobulinemia policlonale può essere associata a stati flogistici cronici o infettivi e altre condizioni non neoplastiche.
Il rilevamento di catene leggere libere nel siero, catene leggere non legate alle Ig è molto importante e un rapporto abnorme κ/λ (intervallo normale da 0,26 a 1,65) indica un eccesso significativo di un solo tipo di catena leggera, espressione della secrezione monoclonale di Ig. Questo esame (FLC) è quindi molto importante in fase diagnostica.
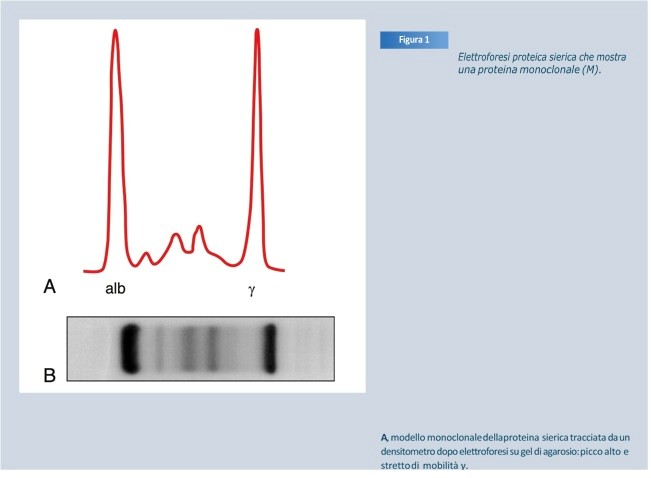
Elettroforesi proteica sierica che mostra una proteina monoclonale (M), modello monoclonale della proteina sierica tracciata da un densitometro dopo elettroforesi su gel di agarosio: picco alto e stretto di mobilità γ (A).
B, modello monoclonale dall’elettroforesi del siero su gel di agarosio (anodo a sinistra): banda densa e localizzata che rappresenta la proteina monoclonale della mobilità γ.
(Da Kyle RA, Katzmann JA. Caratterizzazione immunochimica delle immunoglobuline. In: Rose NR, Conway de Macario E, Folds JD, et al., eds. Manuale di Immunologia di Laboratorio Clinico, 5a ed. Washington, DC: ASM Press; 1997:156, con il permesso dell’American Society for Microbiology)
Vorrei ora soffermarmi sulla gammapatia monoclonale di incerto significato (MGUS) forma più frequente di discrasia plasmacellulare e che richiede un preciso approccio.
GAMMAPATIA MONOCLONALE DI INCERTO SIGNIFICATO (MGUS)
La gammapatia monoclonale di incerto significato (MGUS) è stata per decenni chiamata “Gammapatia monoclonale benigna”. Precisiamo che l’espressione di “Gammopatia” sarebbe scorretta, anche se ormai attestata (30.400 voci individuate dal motore di ricerca di uso più comune), derivando il termine da “patologia delle gammaglobuline” e quindi “Gammapatia” sarebbe il termine corretto (57.200 voci individuate dal motore di ricerca di uso più comune). Certamente poi è più corretta la definizione “di incerto significato” piuttosto che la vecchia definizione di Gammapatia monoclonale benigna, alla luce delle possibili implicazioni della componente M, seppure esigua, in manifestazioni patologiche talvolta anche severe e non necessariamente rappresentate dal MM. Diciamo che il termine “benigna” poteva essere fuorviante relativamente alla necessità di effettuare controlli periodici della discrasia plasmacellulare. La corretta definizione di MGUS è quella di una “alterazione clonale delle plasmacellule con presenza di una proteina M sierica in soggetti che non presentano alcun segno di MM, macroglobulinemia, amiloidosi o altre malattie correlate”.
La caratteristica della MGUS è una concentrazione sierica di proteina M inferiore a 3 g/dL, meno del 10% di plasmacellule nel midollo osseo e assenza di segni clinici o laboratoristici che definiscono invece il MM secondo le linee guida internazionali.
La significatività clinica di una MGUS (espressa dal termine “incerto significato”) è il possibile rischio di trasformazione in MM. Questa probabilità è stata misurata ed è dell’1% all’anno.
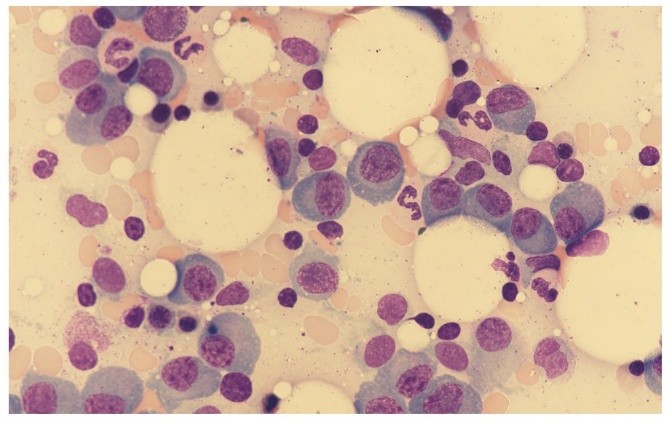
Nella Figura 2 si può vedere un tipico infiltrato plasmacellulare nel midollo osseo.
EPIDEMIOLOGIA
La gran parte dei pazienti in cui viene rilevata una componente monoclonale (M), spesso casualmente (all’esame delle proteine totali e frazionate o protidogramma), ha una MGUS. La prevalenza di MGUS nella popolazione generale aumenta con l’età, si passa da circa l’1% nelle persone di età compresa tra 50 e 60 anni a più del 5% in quelle di età superiore ai 70 anni. Più frequente nel sesso maschile e nella popolazione nera rispetto ai bianchi.
Non è stata mai dimostrata una ereditarietà ma una certa prevalenza di familiarità.
L’etiologia della MGUS è sconosciuta, ma l’età, il sesso maschile, la storia familiare, la soppressione immunitaria e l’esposizione ad alcuni pesticidi sono fattori di rischio noti. Si ipotizza, ma è solo un’ipotesi, che infezioni, stati flogistici e in generale stimoli antigenici, potrebbero determinare anomalie in un clone plasmacellulare e costituire il primum movens della comparsa di questo clone nella maggior parte dei pazienti. Sappiamo bene però che le situazioni appena citate frequentemente e quasi sempre determinano sì una ipergammaglobulinemia, ma questa è quasi sempre policlonale.
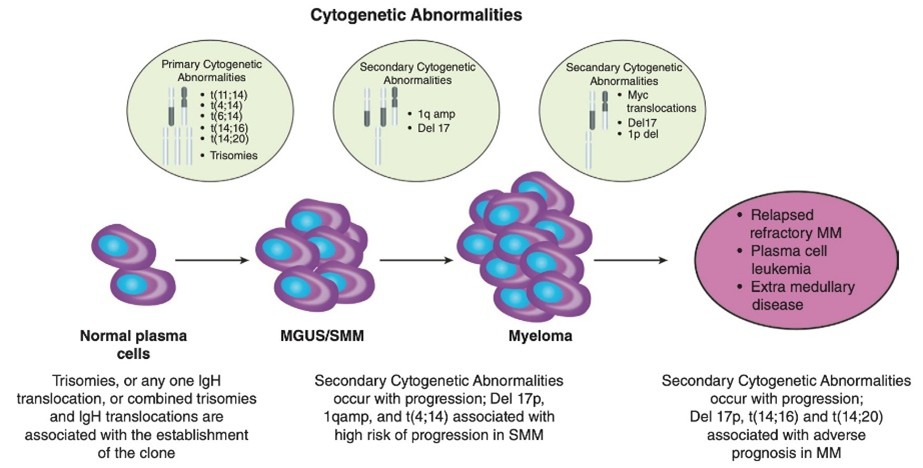
Nelle MGUS possiamo trovare anomali citogenetiche nelle plasmacellule coinvolte. Traslocazioni che interessano il locus della catena pesante della Ig (IgH). Queste sono riscontrabili frequentemente (ca il 40%). Possono essere presenti anche solo trisomie o entrambe le alterazioni e altre anomali genetiche.
Nella Figura 3 si può vedere come anomalie citogenetiche si verificano precocemente, quando la plasmacellula normale passa a uno stadio clonale, definito gammapatia monoclonale di significato indeterminato. Successivamente, si verificano anomalie citogenetiche secondarie associate e che portano a una ulteriore trasformazione. (Da Rajan AM, Rajkumar SV. Interpretazione dei risultati citogenetici nel MM per la pratica clinica. Cancro del sangue J. 2015;5:e365.)
MANIFESTAZIONI CLINICHE
Per definizione le MGUS sono asintomatiche e di solito vengono riscontrate casualmente durante esami di controllo di soggetti sani o affetti da altre patologie per cui si è reso necessario eseguire un protidogramma.
Abbiamo già detto del rischio di evoluzione verso una franca malattia come il MM di circa 1% l’anno per i pazienti con MGUS non-IgM e MGUS a catena leggera; una componente M di tipo IgM potrà evolvere più frequentemente verso la macroglobulinemia di Waldenström. Tutte le forme di MGUS comportano un rischio di progressione all’amiloidosi AL e mi preme subito rammentare come questa possa essere presente anche in presenza di CM minime, oltre che complicare il MM.
Si parlerà di MGUS a seconda del quantitativo di CM, della percentuale di plasmacellule midollari, della presenza o meno di anemia, insufficienza renale, ipercalcemia o lesioni ossee litiche.
Ora l’anemia e l’insufficienza renale sono non infrequenti nella popolazione anziana con MGUS, è pertanto necessario un attento esame dei dati clinici e umorali che ci permetta di mettere in evidenza un danno d’organo dovuto all’infiltrato plasmacellulare e alla secrezione di Ig monoclonale, piuttosto che ad altra causa.
Un dato interessante è rappresentato da quella che è definita “paresi delle Ig” ovvero dall’attenzione da rivolgere ad un dosaggio delle Ig (A, M,G) dove le Ig, diverse dalla Ig monoclonale, siano sensibilmente molto più basse della norma. In pratica: in presenza di CM IgG, trovare IgA e IgM più basse dei valori normali, sarebbe un potenziale segno (criterio minore) di progressione della malattia. Tuttavia dobbiamo anche dire che nel 40% dei pazienti con MGUS potremmo trovare questa “paresi” e quindi certamente l’esame del paziente con MGUS deve essere attento nella valutazione di più parametri clinici e laboratoristici che vanno dalla concentrazione della CM, alla eventuale insufficienza renale legata al danno tipico renale da MM, fino alla presenza di chiari segni di infiltrazione plasmacellulare (con anemia, lesioni litiche ossee e ipercalcemia)
Trattando delle MGUS abbiamo detto che sono asintomatiche per definizione e frequentemente rilevate casualmente. Le caratteristiche di queste “discrasie plasmacellulari” sono rappresentate nel caso delle MGUS da una componente monoclonale (CM) Ig G, A, D, E <3 g/dL con meno del 10% di plasmacellule nel midollo osseo o nel caso di CM IgM meno del 10% di cellule linfoplasmocitoidi nel midollo osseo e CM IgM sempre inferiore a 3 g/dL. Rientrano fra le discrasie plasmacellulari anche quelle forme rappresentate solo da catene leggere sieriche ed alterazione del rapporto k/l (FLC), con numero di plasmacellule sempre <10% nel midollo. Nelle MGUS con CM intera o a catene leggere la proteinuria di Bence Jones deve essere sempre <500 mg/24h. In tutte queste forme saranno poi assenti i segni clinici e laboratoristici di quella che è definita con un acronimo CRAB (“increased Calcium, Renal insufficiency, Anaemia, or Bone lesions”) che indica la presenza di ipercalcemia, disfunzione renale, anemia e lesioni ossee).
Chiaramente, sempre fra le discrasie plasmacellulari, rientrerà poi il classico MM e la cosiddetta forma chiamata “smouldering” che richiede un atteggiamento clinico osservazionale malgrado sia presente una CM superiore ai 3 g/dL e plasmacellule fra il 10 e 60%, sempre però in assenza di CRAB.
Rientrano poi fra le discrasie monoclonali plasmacellulari la macroglobulinemia di Waldenstrom (MW) caratterizzata da CM IgM, presenza di cellule linfoplasmocitoidi ed aspetto clinico assimilabile al linfoma, e altre forme meno frequenti rappresentate dalla amiloidosi AL e dalla sindrome indicata con acronimo POEMS (polineuropatia, organomegalia, endocrinopatia, monoclonalità, lesioni cutanee) e dalle crioglobulinemie etc.
È corretto ricordare infine che la presenza di plasmacellule superiori al 20% in FL e un numero assoluto di 2×109/L plasmacellule nel sangue periferico configurano il quadro della cosiddetta leucemia plasmacellulare.
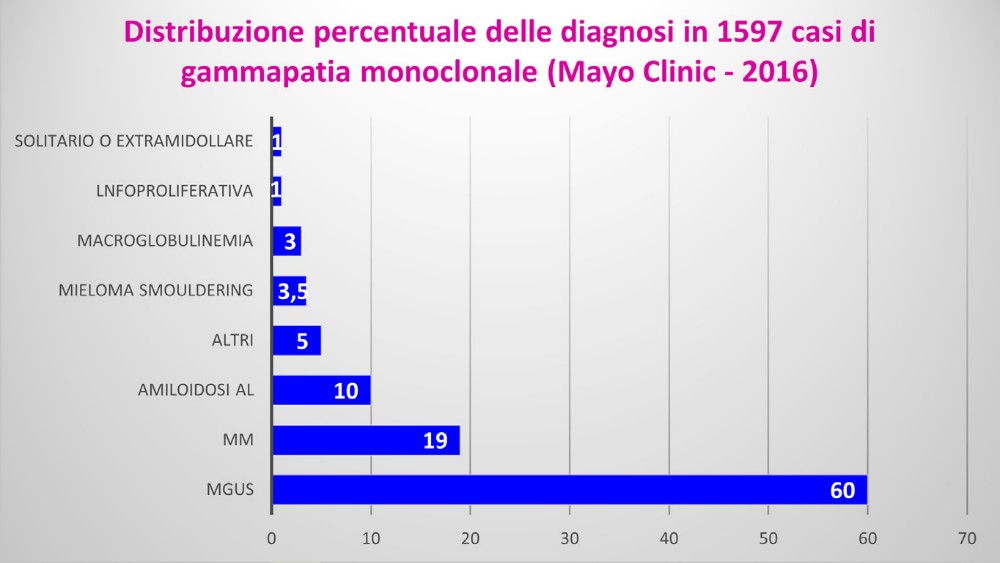
Nella Figura 4 la diagnosi in 1597 pazienti osservati nel 2016 presso la Mayo Clinic con gammapatia monoclonale dove si può notare la maggiore frequenza di MGUS e a seguire MM ed Amiloidosi AL. (dati tratti dal libro “Plasma Cell Disorders” 27^ edizione 2024 Elsevier Inc)
L’elettroforesi e l’immunofissazione del siero e/o dell’urina, o il test FLC sierico vengono eseguiti quando si sospetti MM o WM ed è corretto eseguirli in presenza di sintomi come astenia inspiegabile, aumento importante della VES in assenza di eventi infettivi o flogistici in atto, anemia, dolori ossei, osteoporosi, lesioni o fratture patologiche, ipercalcemia, proteinuria, insufficienza renale o infezioni ricorrenti.
Chiaramente i sintomi sopraelencati non fanno altro che riprendere le situazioni patologiche del MM o della WM ma anche di altre situazioni patologiche associate ad una componente monoclonale. Fra queste vorrei qui ricordare l’amiloidosi AL da sospettare e ricercare in presenza di segni e sintomi di neuropatia periferica neuromotoria inspiegabile, sindrome del tunnel carpale, insufficienza cardiaca congestizia refrattaria, sindrome nefrosica, ipotensione ortostatica, malassorbimento, perdita di peso, cambiamento della lingua o della voce, parestesie, intorpidimento, segni di sanguinamento inspiegabile e steatorrea. Ricordo l’importanza della ricerca dell’amiloidosi AL facendo eseguire un aspirato del grasso periombelicale con colorazione con “rosso congo” e successiva osservazione con microscopio a luce polarizzata (quest’ultima necessaria per la diagnosi). In Figura 5A la colorazione con “rosso congo” del frustolo di grasso periombelicale (A) e l’aspetto all’osservazione a luce polarizzata (colorazione caratteristica “verde-mela”) Figura 5B.

La patogenesi delle amiloidosi è rappresentata dal deposito di fibrille localizzato o sistemico con gravi danni dei tessuti. Nel caso dell’amiloidosi AL gli organi principalmente coinvolti sono cuore e rene.
L’amiloidosi AL è una malattia complessa caratterizzata dal deposito di catene leggere delle immunoglobuline in organi vitali sotto forma di fibrille amiloidi. Queste fibrille da 8 a 10 nm hanno una struttura a foglietto beta pieghettato e sono insolubili. La malattia è progressiva e l’intervento deve essere precoce.
Attualmente non si sa perché si formi l’amiloide AL. Esistono due tipi di amiloidosi AL: una forma localizzata e una forma sistemica. Il primo tipo si riferisce ad una condizione con poca o nessuna immunoglobulina clonale circolante e i depositi di amiloide si creano nel sito delle cellule che producono la proteina clonale. Nell’AL sistemica, c’è un’immunoglobulina clonale circolante, il più delle volte sotto forma di FLC e il deposito di fibrille di amiloide è distante dalle cellule produttrici di immunoglobuline situate nel midollo osseo. L’incidenza dell’amiloidosi AL (precedentemente chiamata primaria) è di circa 12 casi per milione di persone all’anno (valore presumibilmente sottostimato). Circa il 60% dei casi di amiloidosi sono da immunoglobuline monoclonali (AL), circa il 30% sono amiloidosi da transtiretina o TTR, e solo circa il 3% sono amiloidosi secondarie. L’amiloidosi familiare è principalmente quella da transtiretina mutata, ma ci sono anche altre mutazioni rare. L’amiloidosi TTR di tipo selvaggio (wild type) è spesso clinicamente non riconosciuta ed è sicuramente molto più frequente di quanto ci si aspetti. Importante è saper distinguere l’amiloidosi Al dalla TTR per l’approccio terapeutico ovviamente diverso.
In pratica le MGUS vengono diagnosticate casualmente o durante lo studio di una disfunzione renale, neuropatica o di altro organo. Esistono più di cento diverse condizioni mediche associate a MGUS, ma la maggior parte di queste associazioni sono casuali piuttosto che causali.
Ci sono neuropatie associate alla gammapatia monoclonale. Si calcola che circa il 3-5% dei pazienti con gammapatia monoclonale ha una neuropatia periferica e circa il 3-5% dei pazienti con neuropatia periferica ha una proteina M. La neuropatia associata a IgM MGUS si presenta come cronica, neuropatia sensoriale distale, acquisita, demielinizzante, simmetrica. In circa la metà di questi pazienti, la proteina IgM monoclonale riscontrata ha una specificità anti MAG (glicoproteina associata alla mielina).
Al contrario, i pazienti con neuropatia non associata a IgM hanno una presentazione clinica varia. La neuropatia MGUS deve essere differenziata da altri due situazioni sempre legate a discrasia plasmacellulare e che causano neuropatie: l’amiloidosi a catena leggera della Ig (amiloidosi AL) e la neuropatia associata alla sindrome POEMS di cui abbiamo accennato sopra. Evidentemente la presenza di neuropatia associata a CM non può più essere classificata come una MGUS che per definizione è asintomatica.
Introduciamo quindi un concetto che è quello delle gammapatie monoclonali di significato clinico (MGCS) che saranno quelle gammapatie monoclonali che determinano una patologia associata alla presenza di una CM seppure di minima entità. La sindrome POEMS è una di queste condizioni, rara, con sintomi e segni che si riferiscono all’acronimo (POEMS) ed a un altro acronimo associato a questa sindrome, PEST (papilledema, sovraccarico di volume extravascolare, lesioni ossee sclerotiche, trombocitosi ed eritrocitosi). Ci sono poi altre manifestazioni che non trovano collocazione in nessuno dei due acronimi (POEMS e PEST) e sono: livelli sierici elevati di fattore di crescita endoteliale vascolare, ipertensione polmonare, ridotta capacità di diffusione dei polmoni per il monossido di carbonio (DLCO) e tromboembolismo arterioso e venoso. Il sintomo dominante in questa malattia è una neuropatia periferica sensitiva-motoria ascendente. La sopravvivenza complessiva nei pazienti con sindrome di POEMS è molto buona con la terapia diretta come nel MM, verso le plasmacellule, con una sopravvivenza stimata a 10 anni del 79%.
Altre condizioni cliniche legate alla CM sono alcune lesioni renali presenti in queste discrasie plasmacellulari. Oltre i classici danni renali del MM (tubulointerstiziale e glomerulare), esistono svariate glomerulonefriti membrano-proliferative (MPGN) correlate alla sola componente monoclonale delle MGUS. MGUS può causare infatti glomerulonefrite proliferativa per deposizione diretta della proteina M nel mesangio e lungo le pareti capillari, con conseguente glomerulonefrite proliferativa mediata dal complesso immunitario. Può anche causare glomerulonefriti, indirettamente, attivando la via alternativa del complemento con conseguente deposizione di fattori del complemento nei glomeruli, con conseguente glomerulopatia C3. Alla luce di queste lesioni renali in assenza di un MM, è stato coniato Il termine di gammapatia monoclonale di significatività renale (MGRS).
MGRS indica la presenza di una lesione renale che è dovuta ad una gammapatia monoclonale nell’ambito di un disordine proliferativo che non soddisfa i criteri per il mieloma multiplo (MM), la malattia di Waldenström (MW), la leucemia linfatica cronica (LLC) o un linfoma. Bisogna anche precisare che c’è una relazione causale e non casuale fra la gammapatia monoclonale e l’interessamento renale. Sappiamo bene infatti come nella popolazione generale, ma in particolare in età geriatrica, l’insufficienza renale può essere presente insieme ad una MGUS ma spesso casualmente. Nel caso delle MGRS c’è invece un rapporto causa/effetto fra insufficienza renale e MGUS e questo si verifica con una frequenza di circa il 30% nei pazienti con MGUS e IR. Un secondo aspetto da considerare è rappresentato dal fatto che essendo la malattia renale determinata dalla presenza di questa paraproteina, le medesime lesioni renali possono essere riscontrate sia nel contesto di una condizione ematologica benigna (MGRS propriamente detta) sia nel caso di proliferazioni linfocitarie (MW, LLC) o plasmacellulari maligne (MM). La diagnosi di conferma di MGRS richiede l’esecuzione di biopsia renale.
Dopo quanto detto potremmo chiederci: ma lo screening laboratoristico di routine per evidenziare una componente monoclonale si dovrebbe fare?
La risposta attualmente è no, perché non ci sono studi randomizzati per supportare il beneficio di tale strategia. Non ci sono neppure raccomandazioni particolari da offrire ai pazienti a cui viene diagnosticata una MGUS se non periodici controlli nelle MGUS considerate ad alto rischio di progressione e nelle persone più giovani.
Per chi volesse approfondire
- Goldman-Cecil Medicine 27^ edizione 2024 Volume 1 – Cap. 173 S. Vincent Rajkumar “Plasma cell disorders” (173, 1294-1305.e1).
- Rich RR, Fleisher TA, Schroeder Jr. TA, et al. Clinical Immunology Principles and Practice, 6th Edition, December 6, 2022 Elsevier Cap. 79 – Dispenzieri A. Monoclonal Gammopathies (79, 1014-1027).
